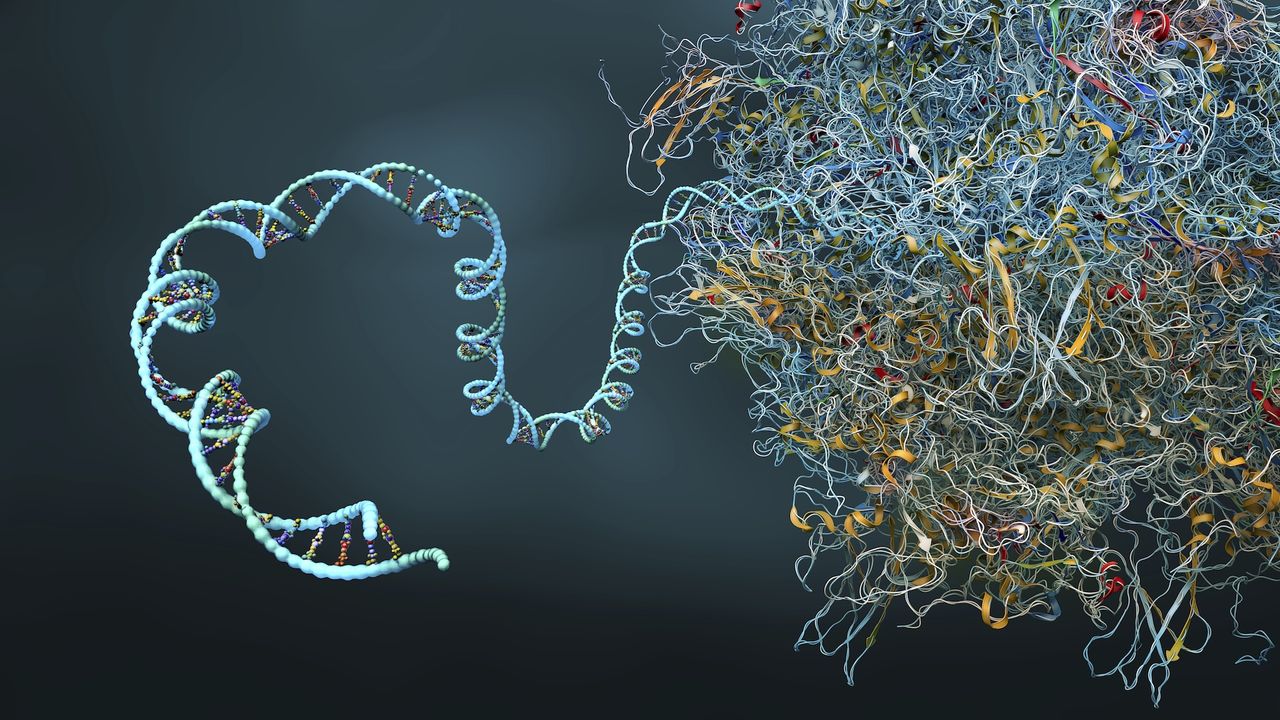
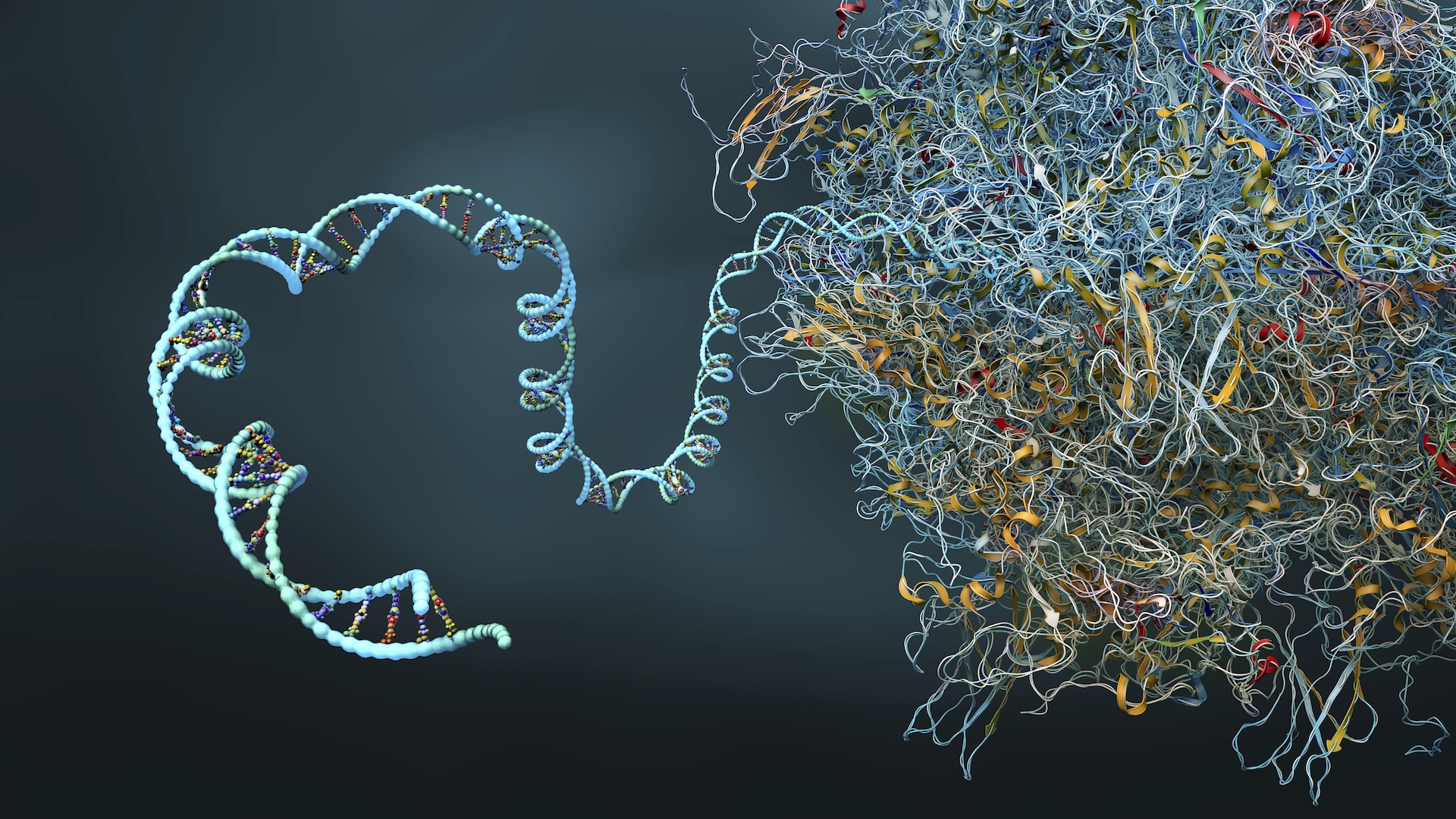
Cercetătorii de la IBM și Moderna au folosit cu succes un algoritm de simulare cuantică pentru a prezice structura proteică secundară complexă a unui 60 de nucleotide pe termen lung mRNA secvență, cea mai lungă simulată pe un computer cuantic.
Acid ribonucleic mesager (ARNm) este o moleculă care transportă informații genetice de la ADN la ribozomi. Direcționează sinteza proteinelor în celule și este obișnuit să Creați vaccinuri eficiente capabil să instigăm răspunsuri imune specifice.
Sale crede pe larg Că toate informațiile necesare pentru ca o proteină să adopte conformația tridimensională corectă este furnizată de secvența sa de aminoacizi sau de „pliere”.
Deși este alcătuit dintr -o singură șuviță de aminoacizi, ARNm are o structură proteică secundară formată dintr -o serie de falduri care oferă o formă 3D specifică a unei molecule. Numărul de permutări posibile de pliere crește exponențial cu fiecare nucleotidă adăugată. Acest lucru face ca provocarea de a prezice ce formă o moleculă de mRNA va lua intractabilă la scări mai mari.
Experimentul IBM și Moderna, conturat într -un studiu Publicat pentru prima dată pentru Conferința Internațională IEEE din 2024 privind calculul și inginerie cuantică, a demonstrat modul în care calcularea cuantică poate fi utilizată pentru a mări metodele tradiționale pentru a face astfel de predicții. În mod tradițional, aceste predicții de obicei s -a bazat pe Calculatoare binare, clasice și inteligenţă artificială (AI) Modele precum Google Alphafold -ul lui Deepmind.
Conform unui nou studiu publicat pe 9 mai pe preprint arxiv Baza de date, algoritmii capabili să funcționeze pe aceste arhitecturi clasice pot prelucra secvențele mRNA cu „sute sau mii de nucleotide”, dar numai prin excluderea unor caracteristici de complexitate mai mare, cum ar fi „pseudoknots”.
Pseudoknots sunt răsuciri complicate și forme în structura secundară a unei molecule care sunt capabile să se angajeze Interacțiuni interne mai complexe decât pliurile obișnuite. Prin excluderea lor, precizia potențială a oricărui model de predicție de pliere a proteinelor este fundamental limitată.
Înțelegerea și prezicerea chiar și a celor mai mici detalii ale pliurilor proteice ale unei molecule de mRNA este intrinsecă pentru a dezvolta predicții mai puternice și, ca urmare, vaccinuri mai eficiente pe bază de ARNm.
Oamenii de știință speră să depășească limitările inerente în Cele mai puternice supercomputere și modele AI prin creșterea experimentelor cu tehnologie cuantică. Cercetătorii au efectuat mai multe experimente folosind algoritmi de simulare cuantică care s -au bazat pe Qubits – echivalentul cuantic al unui bit computer – pentru a modela moleculele.
Inițial folosind doar 80 de qubits (dintr -un posibil 156) pe R2 Heron Unitatea de procesare cuantică (QPU), echipa a folosit un algoritm cuantic variațional bazat pe valoare la risc condiționat (VQA bazat pe CVAR)-Un algoritm de optimizare cuantică modelat după anumite tehnici utilizate pentru a analiza interacțiuni complexe, cum ar fi evitarea coliziunii şi Tehnici de evaluare a riscurilor financiare -Pentru a prezice structura proteică secundară a unei secvențe mRNA de 60 de nucleotide.
Cel mai bun anterior pentru un model de simulare bazat pe cuantic, Conform studiuluia fost o secvență de 42 de nucleotide. Cercetătorii au scalat și experimentul prin aplicarea Tehnici recente de corectare a erorilor pentru a face față cu Zgomot generat de funcțiile cuantice.
În noul studiu de preprint, echipa a demonstrat provizoriu eficacitatea paradigmei experimentale în rularea unor instanțe simulate cu până la 156 de qubits pentru secvențe ARNm de până la 60 de nucleotide. De asemenea, au efectuat cercetări preliminare care demonstrează potențialul de a angaja până la 354 de qubits pentru aceiași algoritmi în setări zgomotoase.
În mod evident, creșterea numărului de qubit -uri utilizate pentru a rula algoritmul, în timp ce scalarea algoritmilor pentru subrutine suplimentare, ar trebui să conducă la simulări mai precise și la capacitatea de a prezice secvențe mai lungi, au spus ei.
Ei au remarcat, însă, că „aceste metode necesită dezvoltarea tehnicilor avansate pentru încorporarea acestor circuite specifice problemei în hardware-ul cuantic existent”, ceea ce indică faptul că vor fi necesare algoritmi și arhitecturi de procesare mai bune pentru a promova cercetarea.