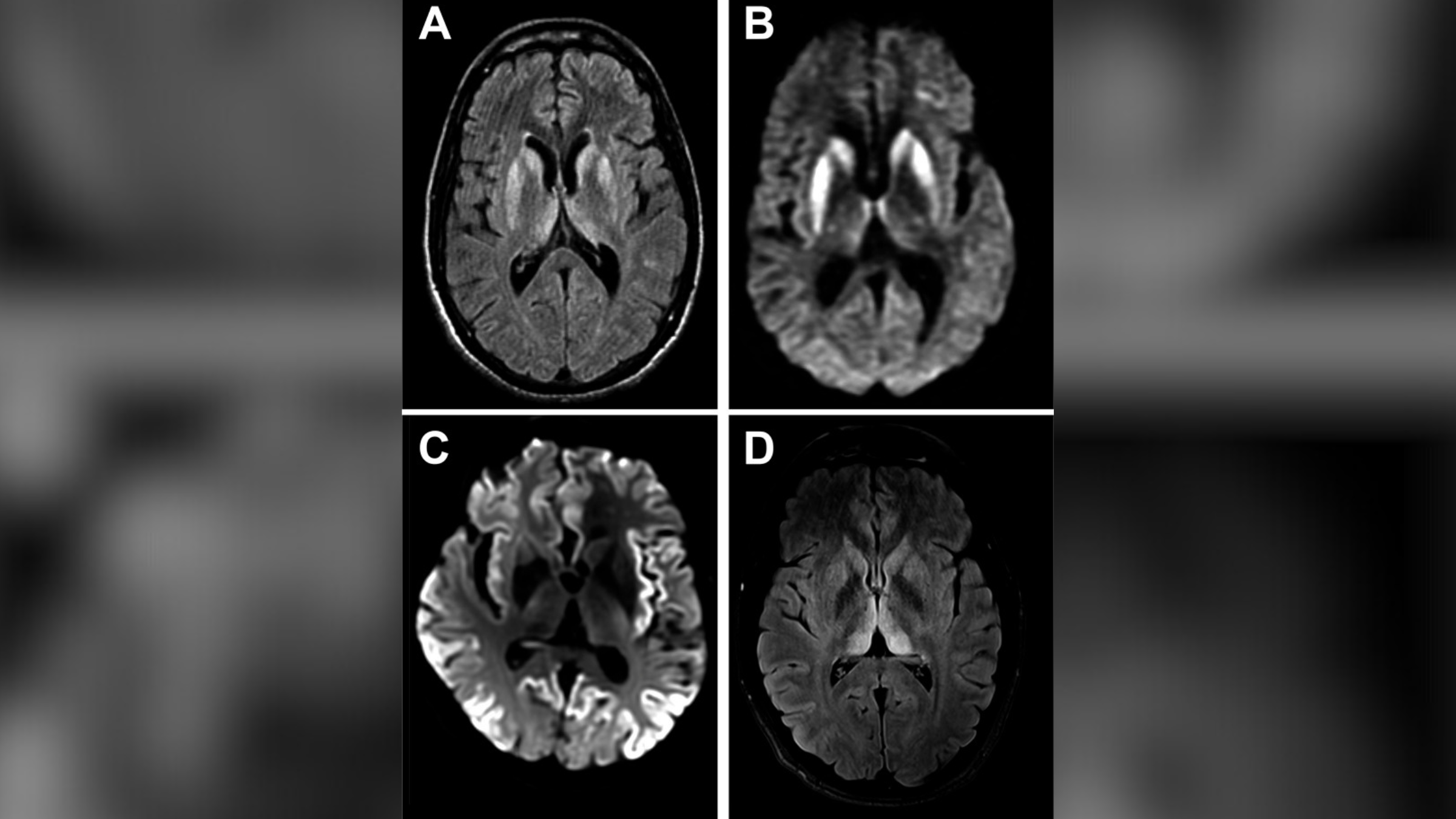
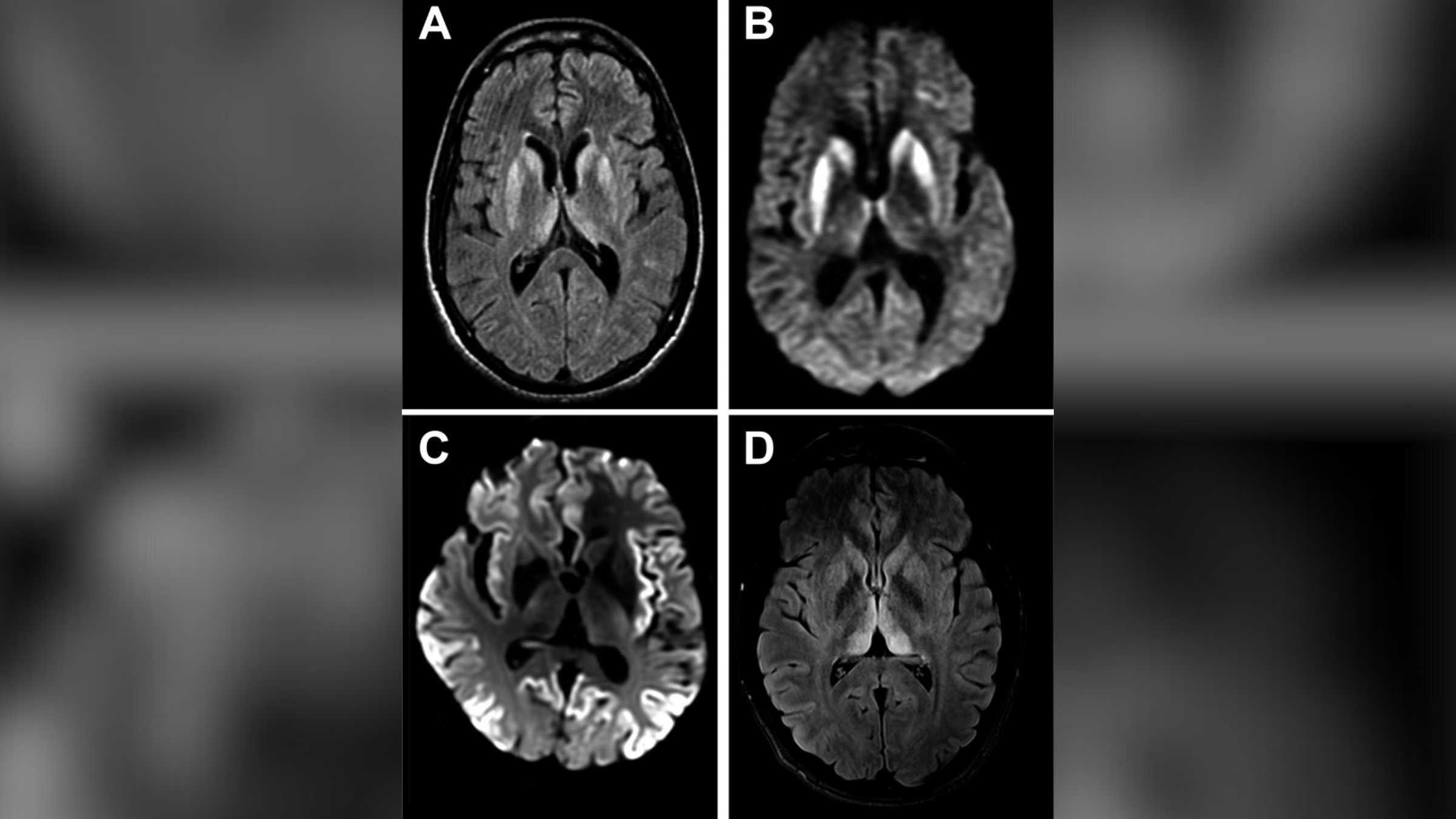
Numele bolii: Boala Creutzfeldt-Jakob (CJD), numită după Hans Creutzfeldt și Alfons Jakob, doi medici germani care au descris prima dată boala în anii 1920.
Populații afectate: CJD afectează în jur de 1 din un milion de oameni din întreaga lume în fiecare an. În SUA, aproximativ 350 de cazuri de CJD sunt diagnosticate anual. Bărbații și femelele sunt la fel de probabil să dezvolte boala.
Cauze: CJD este cauzat de Proteine anormale în creier cunoscut sub numele de „prioni”. Aceste prioni provoacă daune ireversibile țesuturilor, ceea ce duce la formarea de găuri asemănătoare cu burete în întregul creier care provoacă neuroni a funcționa defectuos și a muri. Prionii stârnesc o reacție în lanț, ceea ce a determinat altul, Proteine normale din creier pentru a -l înșela greșit. Acest lucru perpetuează starea și determină pacienții să se experimenteze progresiv mai rău Probleme cu mișcare și funcție mentală.
Înrudite: LAB Tech dezvoltă starea fatală a creierului după accident cu mostre „boli de vacă nebună”
Există trei tipuri principale de CJD, care diferă în funcție de modul în care prionii își au originea în creier. Cea mai frecventă dintre aceste forme este „CJD sporadic”, care reprezintă Aproximativ 85% din cazuri. CJD sporadic apare atunci când proteinele normale au greșit spontan și devin prioni din motive necunoscute, simptomele de obicei se dezvoltă mai întâi la adulții care sunt Între 45 și 75 ani.
În plus, între 10% și 15% din cazuri de CJD sunt cauzate de o mutație într -o genă numită PRNP, ceea ce duce la dezvoltarea de prioni. Această formă genetică de CJD este moștenită într -un mod dominant autosomalceea ce înseamnă că un copil nu trebuie decât să moștenească o copie a genei defecte de la oricare dintre părinte pentru a dezvolta afecțiunea. CJD genetic apare cel mai adesea la oameni Între 30 și 50 de ani.
Mai puțin de 1% din cazurile CJD sunt „infecțioase”, ceea ce înseamnă că sunt declanșate de transmiterea prioniilor din surse externe. O modalitate prin care se poate întâmpla acest lucru este atunci când oamenii mănâncă carne de vită de la vaci care au encefalopatie spongiformă bovină, mai cunoscută sub numele de „boală de vacă nebună”. SUA au avut reglementări stricte din anii ’90 pentru a împiedica acest lucru să se întâmple. De la descoperirea din 1996, că oamenii ar putea primi CJD de la „vacile nebune”, ” Doar 233 astfel de cazuri au fost raportate la nivel mondial.
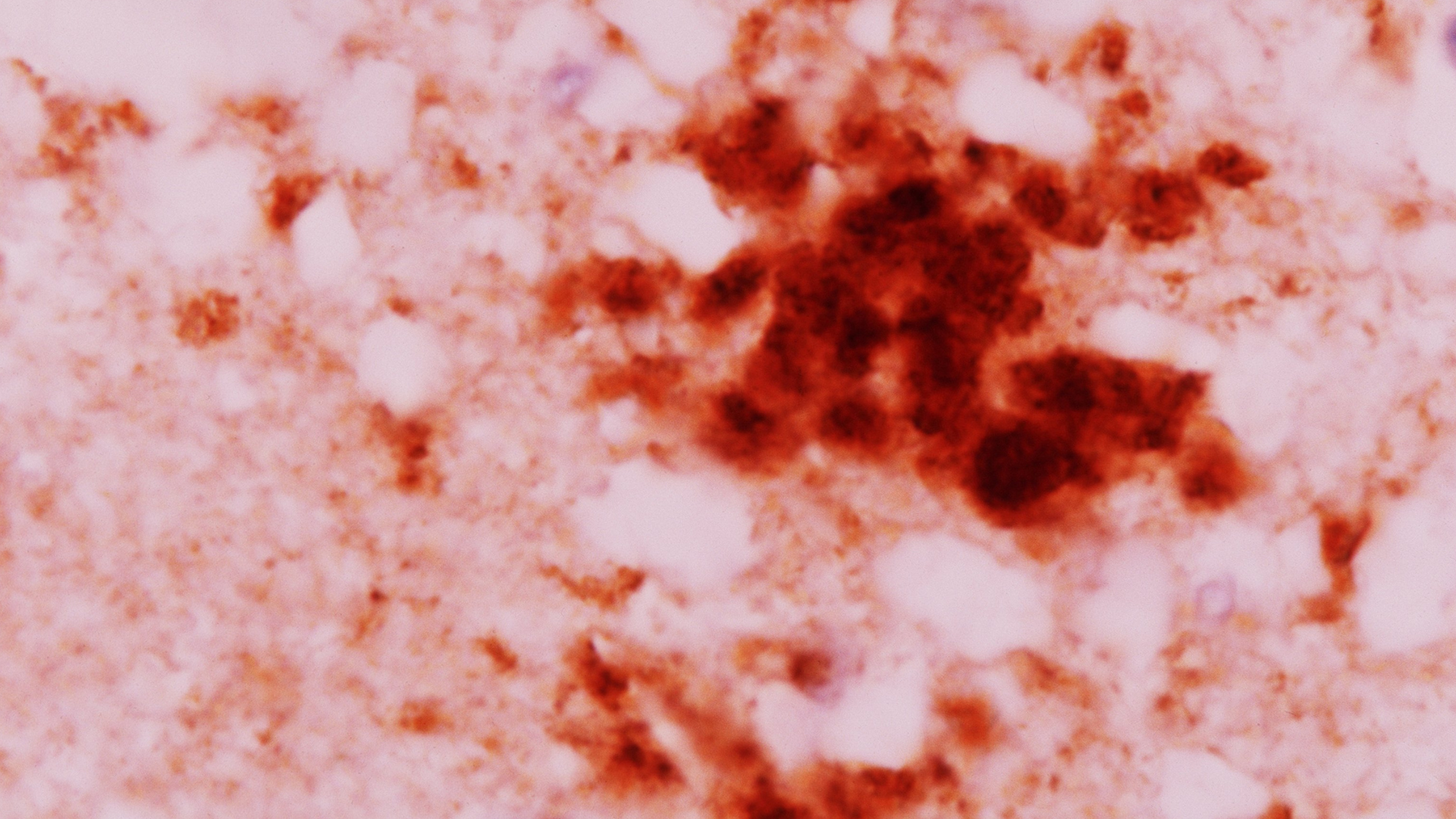
Un alt mod în care CJD poate fi transmis este atunci când este trecut accidental de la om la om în timpul procedurilor medicale – de exemplu, dacă un pacient primește un transplant sau o transfuzie de sânge de la un donator cu CJD. Un exemplu proeminent în acest sens s -a întâmplat de la sfârșitul anilor 1950 până în 1985când medicii au dat pacienților hormoni de creștere contaminați derivați din cadavre. Acest lucru a dus la Cel puțin 226 de cazuri de CJD la nivel mondialinclusiv 29 de cazuri în SUA
Simptome: Simptome comune ale CJD Includeți demență, confuzie și dezorientare, halucinații, lipsa de coordonare și rigiditatea musculară. De asemenea, pacienții pot experimenta schimbări de personalitate, somnolență, convulsii și probleme care vorbesc. În plus, pot avea simptome psihologice, cum ar fi ca depresie severă, anxietate și iritabilitate.
Simptomele CJD progresează adesea rapid până la punctul în care pacienții devin complet în pat, neștiind de împrejurimile lor și nu pot comunica cu cei din jurul lor. CJD este întotdeauna fatalși Aproximativ 70% dintre pacienți mor într -un an de diagnostic, cel mai adesea din cauza o infecție, o eroare a inimii sau a pulmonarului.
Tratamente: Nu există leac pentru CJD, ci droguri poate ajuta la ameliorarea simptomelor unui pacient. De exemplu, pacienților li se poate prescris medicamente pentru a reduce smucitura mușchilor sau pentru a atenua anxietatea.
Diagnosticul precoce al formei genetice a CJD poate ajuta pacienții, permițându -le Faceți aranjamente pentru sfârșitul îngrijirii vieții și planificarea familiei.
Renunțare
Acest articol este doar în scop informativ și nu este menit să ofere sfaturi medicale.
Mai multe despre boala infecțiilor cu viruși